



The World Health Organization (WHO) defines clinical trials as a type of research that studies new tests and treatments and evaluates their effects on human health outcomes (1). Clinical trials play a pivotal role for the approval and marketing authorization of new drugs, medical devices, and biologicals by regulatory bodies globally. Studies in humans most accurately reflect both harms and benefits of the drug unlike animal studies and computer-based drug discovery methods. In addition, clinical trials are often the longest and most costly phase in the drug development program making it essential to design and conduct trials with utmost care keeping economic and time considerations in mind for the company. The clinical development program in most countries, including Australia is divided into four phases as shown below:
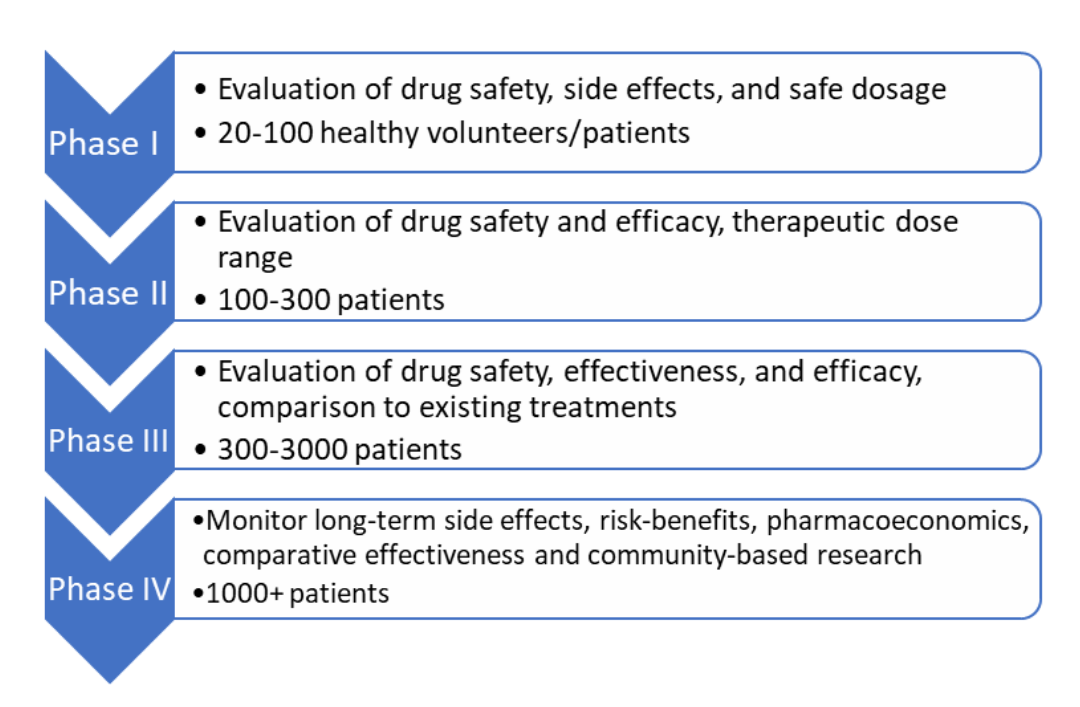
Phase 1 to 4 clinical trial service step by step process
Australia is often looked upon by foreign sponsors as a suitable location for clinical trials owing to several benefits from an infrastructure and regulatory perspective. World-class infrastructure and health professionals, an efficient regulatory scheme, government incentives for foreign sponsors, accessibility to a culturally diverse population, and high-quality research facilities have made Australia a ‘hot spot’ for clinical trials. The rising popularity of Australia can be seen by a 6% p.a. increase in trials from 2015 to 2019 recorded in the Australian New Zealand Clinical Trials Registry (ANZCTR). (2). Therefore, it is essential for sponsors to have a clear and in-depth understanding of the process of clinical trial approval in Australia.
Who regulates clinical trials in Australia?
The Therapeutic Goods Administration (TGA) which is a part of the Australian Government Department of Health is responsible for regulating therapeutic goods including prescription medicines, biologicals, dietary supplements, and blood and blood products. The Australian Register of Therapeutic Goods (ARTG) is a repository of all products with therapeutic claims.
Ethical Considerations for clinical trials in Australia
Keeping patient safety and well-being at the forefront of all clinical operations, trials in Australia for ‘unapproved’ therapeutic goods should be conducted in accordance with the requirements of the therapeutic goods legislation, Declaration of Helsinki principles, the National Statement on the Ethical Conduct of Human Research, Good Clinical Practice (GCP) guidelines, and other site-specific requirements (3). ‘Unapproved’ therapeutic goods are any medicine, medical device, or biological not included in the Australian Register of Therapeutic Goods (ARTG). In addition to global GCP guidelines, the National Statement on the Ethical Conduct of Human Research which has been developed by the National Health and Medical Research Council (NHMRC) should be adhered to for conducting research in an ethical manner . Since ethics committees have the primary role of assessing and approving trial proposals and trial monitoring, compliance with the ethical statement is of utmost importance.
How is a clinical trial for ‘unapproved’ therapeutic goods approved in Australia?
The clinical trial approval process for ‘unapproved’ therapeutic goods in Australia consists of the following phases:
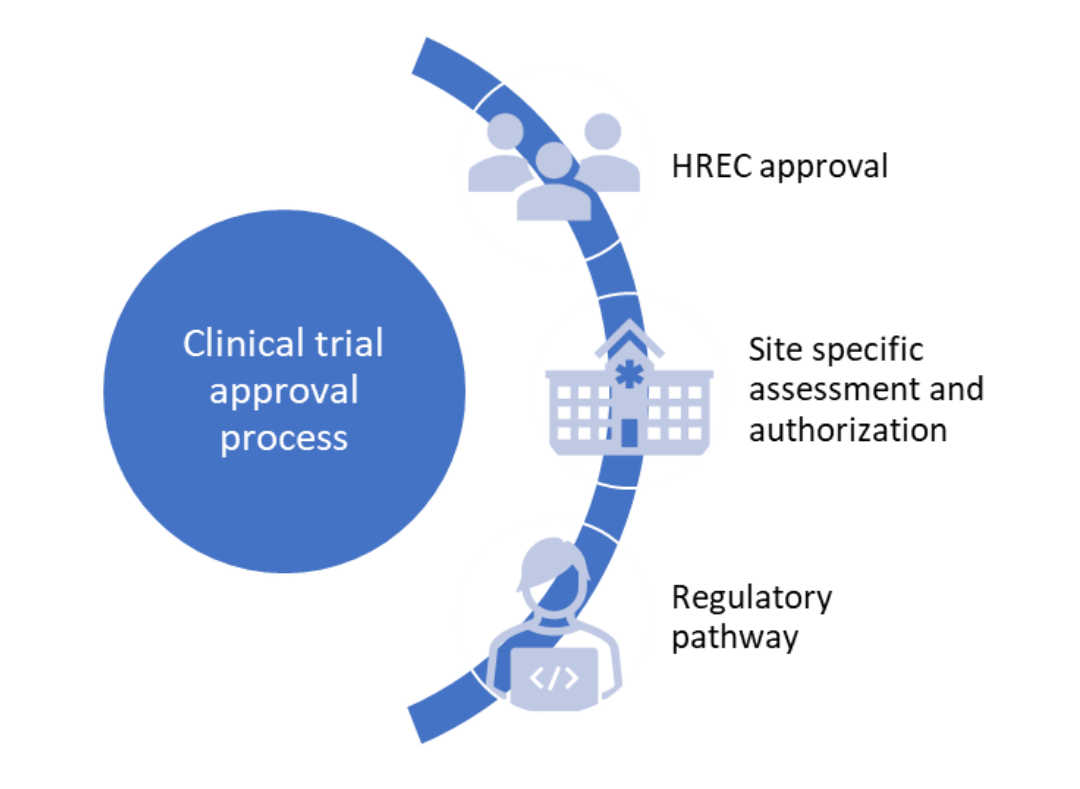
How is a clinical trial for ‘unapproved’ therapeutic goods approved in Australia?
Human Research Ethics Committee (HREC) approval
Unlike the United States and European Union, where regulatory approval from the FDA or appropriate regulatory authority is necessary to start clinical trials, the HREC is responsible for reviewing the scientific and ethical aspect of clinical trial proposals in Australia. Regular monitoring of the clinical trial progress and determination of the type of regulatory pathway required for approval are other functions of the HREC. It is necessary for the ethics committee to comply with the guidelines issued by the NHMRC regarding their constitution and operation and should be registered with the NHMRC.
HRECs are responsible for reviewing the following documents:
- Trial protocol
- Investigator’s brochure
- Informed consent forms
- Patient information documents
- Subject recruitment plans
- Planned payment or compensation initiatives
- Investigator(s) qualifications
- Additional information, if required
In addition to trial approval, the HREC is also responsible for ongoing review of trials which is dependent on subject risk. The HREC should be notified by the investigator(s) of any deviations from trial protocol to prevent subject harm. Adverse drug reactions, safety information, and changes that can affect the risk/benefit of the trial should be communicated to the HREC. Withdrawal of approval of the trial by the HREC must result in stoppage of trial activities.
Application for ethics approval from HREC involves the following steps:
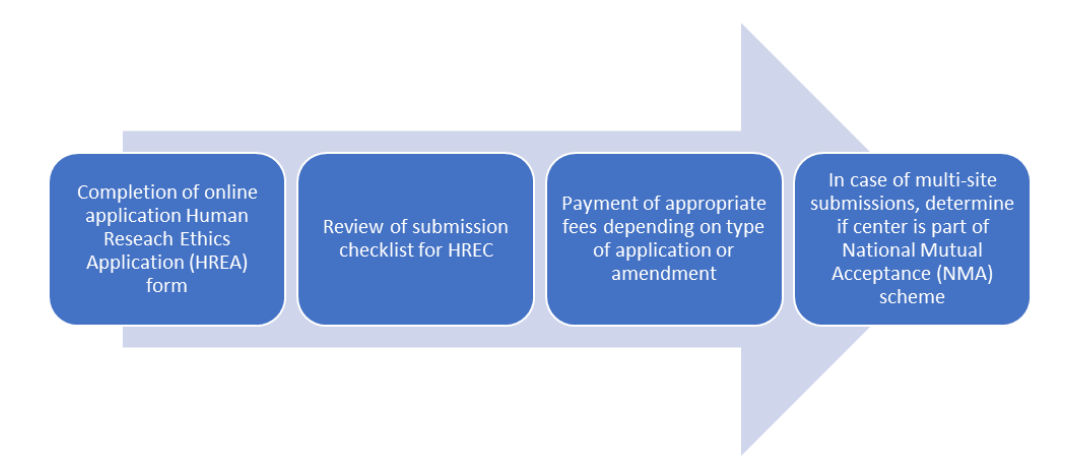
Application for ethics approval from HREC involves the following steps:
Site specific assessment and authorization
The institution or organization (approving authority) where clinical trials are carried out (trial sites) are responsible for site assessment and authorization processes which is a part of the research governance process. Assessment of the site for adequate resources, facilities, and infrastructure forms the core of the site assessment process. The final decision to conduct a trial at a particular site lies with the approving authority despite ethical approval by the HREC. Various steps are necessary to determine ‘site readiness’ for a clinical trial and include Feasibility assessment: which involves the determination of alignment of the proposed trial with the institutional mission, values, and priorities. Various factors areas that are assessed include the availability of resources such as infrastructure, capacity, and participant population and recruitment within a given budget.

- Document preparation and submission: completion of site-specific assessment (SSA) form by the local site principal investigator (PI) or site coordinator. Separate SSA forms need to be completed for each site by the PI in case of multi-center research. The SSA form contains information such as:
- Site name
- Name of lead HREC
- Project title
- Type of research
- NHMRC research group
- Information about site PI/associate investigators
- Project contact person
- Project description
- Project start and finish dates
- Departments/services to be involved in research
- Research personnel expertize
- Participant recruitment details
- Access to information and data
- Intellectual property
- Clinical trials information
- Insurance and indemnity
- Biosafety, chemical, and radiation safety
- Investigational drugs
- Funding
- Financial oversight
- Other support
- Site assessment and authorization: The site assessment documents are reviewed by the research governance officer (RGO) at the site for endorsement. Following endorsement, the application is forwarded to the Chief Executive (CEO)/Director of the site for final consideration and authorization endorsement by the RGO and authorization by the CEO should be communicated to the PI followed by registration with the clinical trials registry.
Regulatory pathway
There are two schemes under which clinical trials involving ‘unapproved’ therapeutic good are carried out in Australia, the Clinical Trial Notification (CTN) and the Clinical Trial Approval (CTA, previously known as CTX) scheme. The sponsor and HREC determine the type of scheme to be used depending upon the availability of data which in turn dictates the level of involvement of the TGA (4,5).

CTN Scheme
The process for application for the CTN and CTA schemes is shown below:
CTN scheme
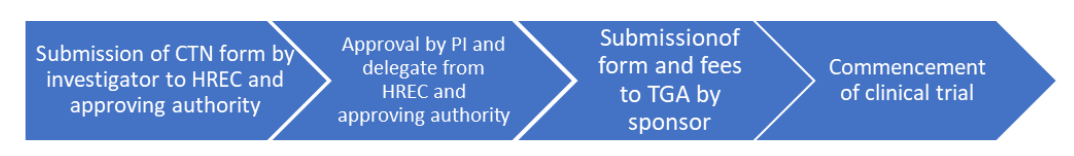
CTN scheme (notification scheme)
CTA scheme

CTA scheme (evaluation scheme)
The TGA should be notified of any changes to the trial protocol and should be informed when a trial is completed at any site. The world-wide acceptability of trial data from clinical trials conducted in Australia is largely due to its strict ethics approval system and high-quality research facilities. Sponsors generally choose Australia as a clinical trial destination based on an efficient regulatory system that saves time.
Read More:
Advantages of Conducting Clinical Trials in Australia
Need Support to Conduct your Clinical Trials in Australia?
We’d be delighted to assist you in conducting your clinical trials in Australia
Connect with our experienced staff in Australia today!!
Provide details of your requirements by clicking on the given link below and our professional team of clinical research experts will get in touch with you right away.
References
- Clinical trials (who.int)
- Australia accounts for 3.9% share of global clinical trial activity in 2020 – Clinical Trials Arena
- Australian clinical trial handbook. Guidance on conducting clinical trials in Australia using ‘unapproved’ therapeutic goods. Version 2.4, August 2021, TGA Health Safety Regulation
- Access to unapproved therapeutic goods – Clinical trials in Australia | Therapeutic Goods Administration (TGA)
- Clinical Trials Sector Reports : MTPConnect

