



As per Section 201(h)(1) of the Food, Drug, and Cosmetic Act, a device is an instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including a component part, or accessory which is recognized in the National Formulary or United States Pharmacopoeia, intended for use in the diagnosis, mitigation, cure, prevention, or treatment of a disease, or intended to exert its effect without chemical action. In the United States, the Food and Drug Administration (FDA) regulates the approval and marketing of medical devices subject to regulatory controls in the Federal Food, Drug, and Cosmetic (FD&C) Act and regulations in Title 21-Code of Federal Regulations (21 CFR)
Parts 1-58. Since there are several classes of devices with different risk profiles, it is important for the Sponsor/investigator to select an appropriate application pathway for their device to obtain marketing authorization.
The FDA recommends a stepwise process to understand the device and select a suitable regulatory pathway as follows (Figure 1):
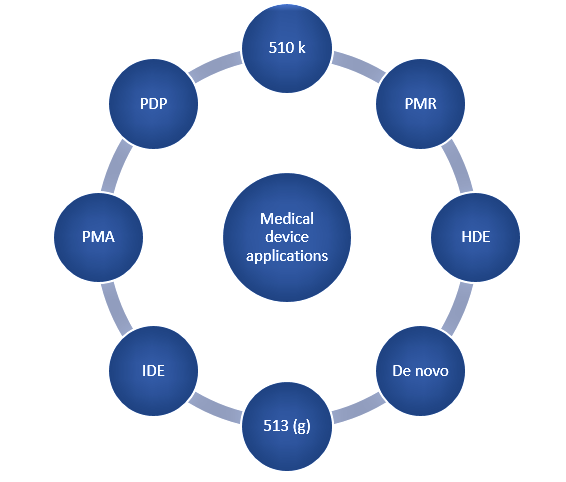
Figure 1. Steps for marketing of medical devices as per US FDA
The most important factor is the correct classification for the medical device (Step 1). The US FDA categorizes medical devices into three classes depending on the degree of risk and accompanying regulatory controls.
Class I
These devices present the lowest risk and therefore minimal potential for harm. They are subject to the least stringent controls. Some Examples include enema kits, elastic bandages, handheld surgical instruments, nonelectric wheelchairs.
Class II
These devices present a moderate risk and their potential for harm is greater than for Class I devices. Some examples include CT scanners, infusion pumps for intravenous medications, powered wheelchairs, pregnancy test kits.
Class III devices:
These devices present the highest risk and sustain, or support life and are implanted. They present potential unreasonable risk of illness or injury and are subject to the strictest regulatory controls. Some examples include breast implants, pacemakers, defibrillators, cochlear implants, implanted prosthetics.
Once the device is classified, the Sponsor can go on to identify the correct premarket submission pathway. The FDA website has a product classification database that helps in the selection of an appropriate submission type based on the product’s classification. The various pathways that are commonly used are depicted in Figure 2 and explained in detail below.
510 (k) application:
This is a premarket submission made to the FDA to demonstrate that the device to be marketed is safe and effective and substantially equivalent to a legally marketed device or predicate device in terms of intended use, technological characteristics, and performance testing. Some class I and most class II devices require a 510 (k) unless they are exempt. A 510(k) submission includes a detailed device description, labeling information, demonstration of substantial equivalency, and supporting data from performance testing.
Premarket approval (PMA):
This is the process for scientific and regulatory review to evaluate the safety and effectiveness of Class III medical devices and is the most stringent process. The devices for which this approval is required generally involve new concepts and are not of a prior type. PMA submissions take longer to secure and require more detailed information such as information from clinical trials. Other details that must be included in the submission is information of the device’s safety and effectiveness, description of the device’s design, components, and manufacturing process, data from preclinical and clinical studies, labelinginformation, and summary of safety and effectiveness.
513(g) application:
This is a request for classification information from the FDA and is a reference to section 513 (g) of the FD&C Act. This enables the manufacturers to seek the views of the FDA on their device classification and obtain information on requirements that apply to their device such as PMA or 510 (k). The FDA charges a fee for this service from the manufacturers and the application should include a detailed description of the device, proposed labeling and uses, and marketing material to make their decision. This process if valuable for manufacturers and decreases the chances of making errors in choosing a suitable regulatory pathway.
Investigational Device Exemption (IDE):
This is like an Investigational New Drug (IND) application and allows the investigational device to be used in clinical trials to support PMA for class III and some class II devices. Unless the device is exempt, all investigational devices require an IDE prior to use in humans. An IDE submission must include sponsor information, report of prior investigations, investigational plan, manufacturing information, certification by investigators, institutional review board (IRB) information, conducting investigator site information, environmental assessment forms, informed consent forms.
Product Development Protocol (PDP):
This is a process for market approval of class III devices that have a well-established technology. In this method, the clinical evaluation of the device and necessary requirements for marketing approval are submitted in one document. It allows the sponsor to interact with the FDA regarding expectations for demonstration of safety and efficacy of a new device and ensures predictability once the agreement is made. The PDP contains information about design and development activities and their output and acceptance criteria for which milestones are submitted to the FDA. A PDP declared complete by the FDA is an approved PMA.
De novo application:
This risk-based classification process is suitable for novel class I and II devices for which there is no predicate available and which are demonstrated to have a low to moderate risk. In this case marketing approval for a device is based on general and special controls to provide assurance of safety and effectiveness. Devices established as de novo can be used as predicate devices in future 510 (k) applications. The submission includes administrative information, regulatory history, device description, indications, classification summary and recommendations, clinical, nonclinical, and biocompatibility information, benefit-risk consideration and mitigation strategies, and labeling information. Unlike 510 (k) applications which require a predicate for demonstration of substantial equivalence, de novo device applications may require clinical studies as no predicate is available.
Post market requirements (PMR):
This is required for devices already on the market and include post market surveillance (PMS) studies and post approval studies at the time of approval of PMA or PDP applications. PM studies involve gathering user data from real world studies and analyzing it to make necessary changes or recommendations on the device. PMS is done through the MedWatch system in the United States which allows users of medical devices to submit information on adverse events associated with device use. PMS plans are most important for class II and III devices that are for pediatric use, implantable, or life-sustaining or life-saving devices for which it is mandatory for the sponsor/manufacturer to submit a PMS plan including objective, study design, proposed schedule, and reports.
Humanitarian Device Exemption (HDE):
This is a marketing application for a humanitarian use device (HUD) which is a medical device intended to benefit patients in the treatment or diagnosis of a disease or condition that affects small or rare patient populations. It is necessary for the device to be classified as a HUD by the FDA following which a HDE application is made. The application is like a PMA and should contain a risk-benefit profile and ensure that no comparable device is available on the market. However, it is not necessary for the HDE to have clinical information unlike a PMA as sufficient patients may not be available to conduct a trial.
As different classes of medical devices are available, it is important for sponsors and manufacturers to understand the regulatory pathway and marketing application that should be made. This will allow the maximum chances of success obtaining marketing approval. The FDA also has facilities for interaction with the agency that provide guidance on the content, submission, and review of applications in a timely manner and should be utilized by sponsors.
References
2) How to Study and Market Your Device | FDA
3) Premarket Notification 510(k) | FDA
